
OpenFold3 is finally out! Before diving into the what this means for the future of protein structure prediction, let's see what effect the release will have today. OpenFold3 is a fully open source alternative AlphaFold3, made with the backing of the OpenFold Consortium, a collection of biotech and pharma companies, which Tamarind Bio is a part of. Let's look at the benchmarks.
Where are we now?
As with other commercially accessible AF3 reproductions, OpenFold3 does lag behind in some modalities/applications and meets/beats AlphaFold3 in others.
Small-molecule (docking/poses)
OF3 roughly matches AF3 for ligands somewhat similar to the training data, trailing AF3 on the hardest novelty ligand categories (e.g., GUNS & Poses)
Training cutoff matches AF3 for fairness; some competitors use newer data, making plots not directly comparable.
Antibody–antigen
Large gap: AF2 remains the best in benchmarks for antibody-antigen docking against Boltz, Chai, and OpenFold3 according to our benchmarks, all of these trail AF3 substantially
AF3 benefits from larger amounts of sampling compared to alternatives, OpenFold is no exception
Monomers & multimers
All AlphaFold-style structure predictors roughly converge on the same accuracy for single chain proteins and non-antibody complexes, with a slight edge towards AF3.
RNA
Best modality for OF3: at/above AF3 on shown metrics.
Likely win traced to data prep: during cropping, count only polymers toward the 20-chain budget (don’t count ions/small ligands), preserving RNA context without oversampling.
Where do we go from here?
Outside of the highest accuracy result reached in RNA structure prediction, this preview release of OpenFold3 follows expected results, but presents a template for what the next generation of biomolecule structure predictors might look like. Some concrete things would be achieving parity with AF3, which would entail a full re-training with the whole of the PDB. Other applications might include Boltz-2 style protein-ligand binding affinity predictions, conformational ensembles, fast inference for virtual screening.
One less obvious hurdle to overcome, especially for antibodies, is matching AF3's increased efficacy with additional sampling. The DeepMind team find that producing a very large volume of structures for the same inputs (e.g. 100 to 1,000 samples) and picking the best one substantially increases the accuracy of the docked poses produced. OpenFold3 (same with Chai or Boltz), don't currently show these returns.
Now, for some speculation on the next generation of structure predictors:
Physics is likely to have a greater role in reasoning. ML models are good at capturing evolutionary clues, but this does not help when that information isn't represented in training data. Rigid, well-constrained regions are largely solved; the hard parts are flexible, functional segments. We'll certainly see some diffusion components that steer exploration through structure space, MD-inspired objectives, and local energy minimization for cleanup.
Multiple sequence alignments seem to persist despite language-model based approaches, and they aren’t disappearing so much as evolving. While large single-sequence models begin to internalize MSA-like statistics, the near-term sweet spot is smaller models coupled to retrieval, bringing MSA in at inference rather than relying on giant monolithic memorization. Calibration will also improve by training on explicit negatives (non-interacting pairs and disordered regions) to curb false-positive PPIs and overconfident helices where disorder is expected, so confidence reflects biochemical reality rather than assuming an input is a protein/stable complex.
Using OpenFold3 on Tamarind Bio
To leverage OpenFold3's capabilities, a researcher could follow this streamlined workflow on Tamarind:
Access the Platform: Begin by logging in to the tamarind.bio website.
Select OpenFold3: From the home screen, search for and choose the OpenFold3 tool tile, which will bring you to the OpenFold3 application.
Input Sequences: Select from the 4 input options, whether you want to input a Protein, RNA, DNA, or Ligand sequence. (Optional, select your Output Format from either mmCIF or PDB)

Run Prediction: The platform accesses the NVIDIA NIM Microservice for accelerated inference.
Analyze the Prediction: The model outputs the predicted structure with its confidence scores (pLDDT, pTM). From here, you can interact with the molecule, looking at specific areas of interest. Below, you can see the chart showing different values from your run, which you can analyze, compare/contract, etc utilizing an export csv function or by utilizing our AI Copilot to run bulk analysis for you via the "Analyze CSV" button.
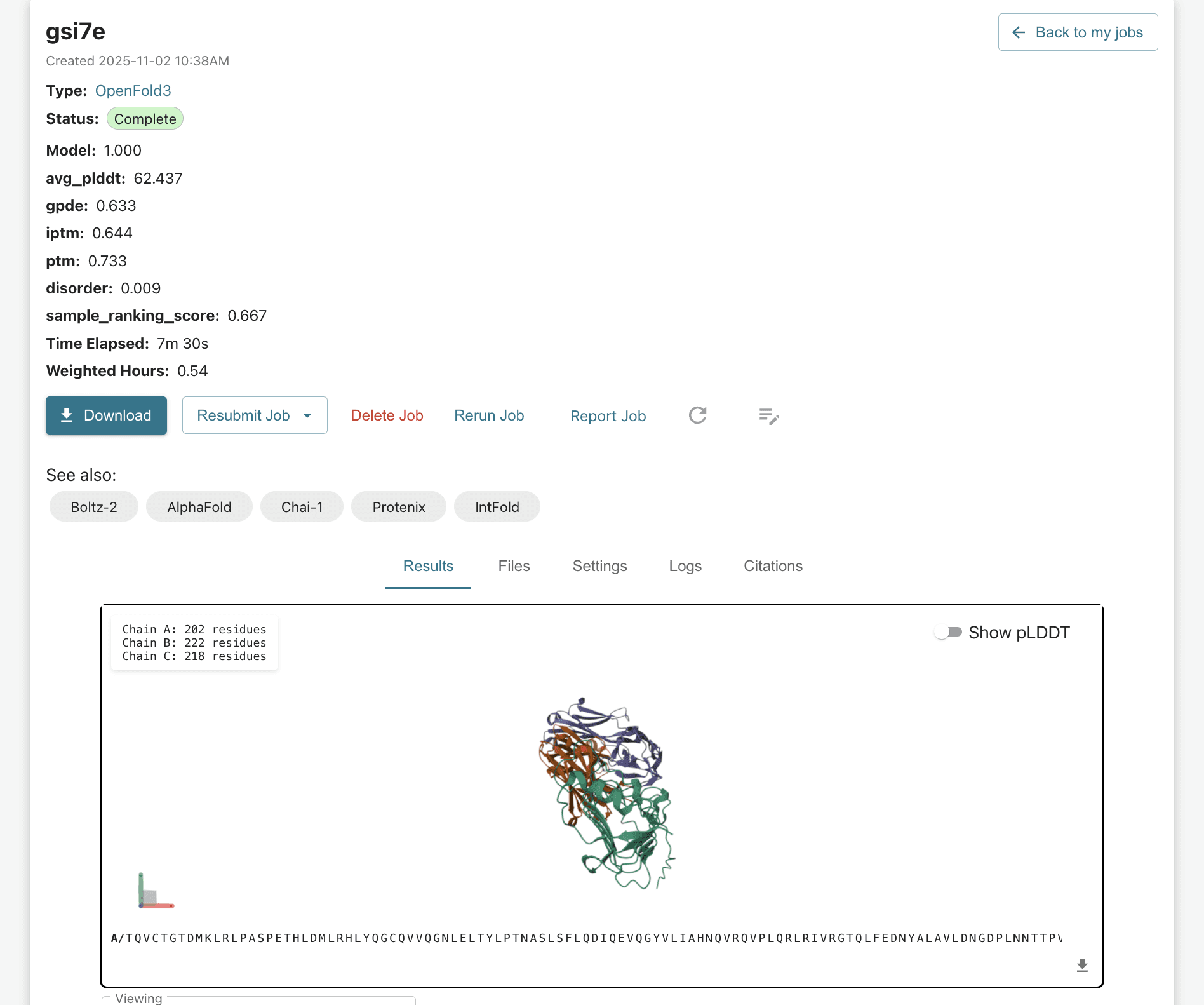
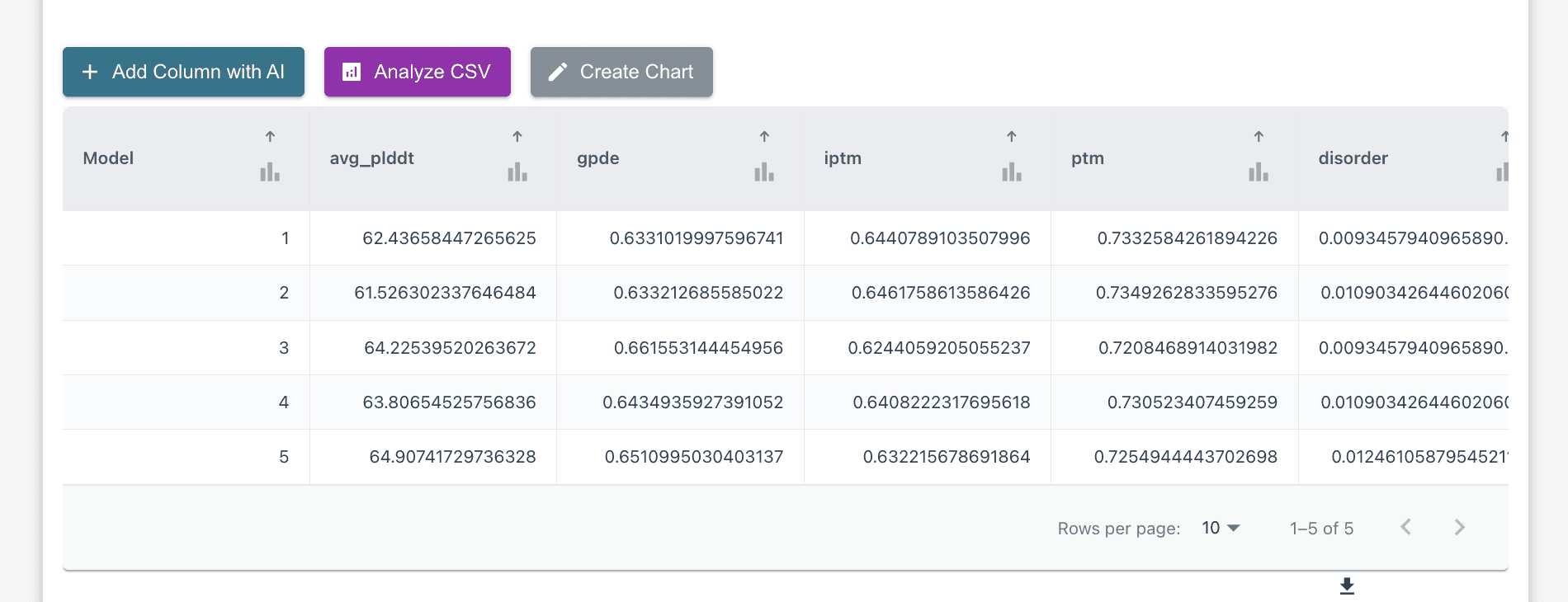
Prioritize Candidates: Researchers can use the predicted confidence scores to prioritize structures for further study, leveraging the model's distinct failure modes (which are complementary to AF3) for comprehensive screening.
To learn more about the OpenFold3 project, check out the OpenFold consortium's website here.